Autosomal Recessive Polycystic Kidney Disease (ARPKD)
Morphological and Histopathological findings
Image Description
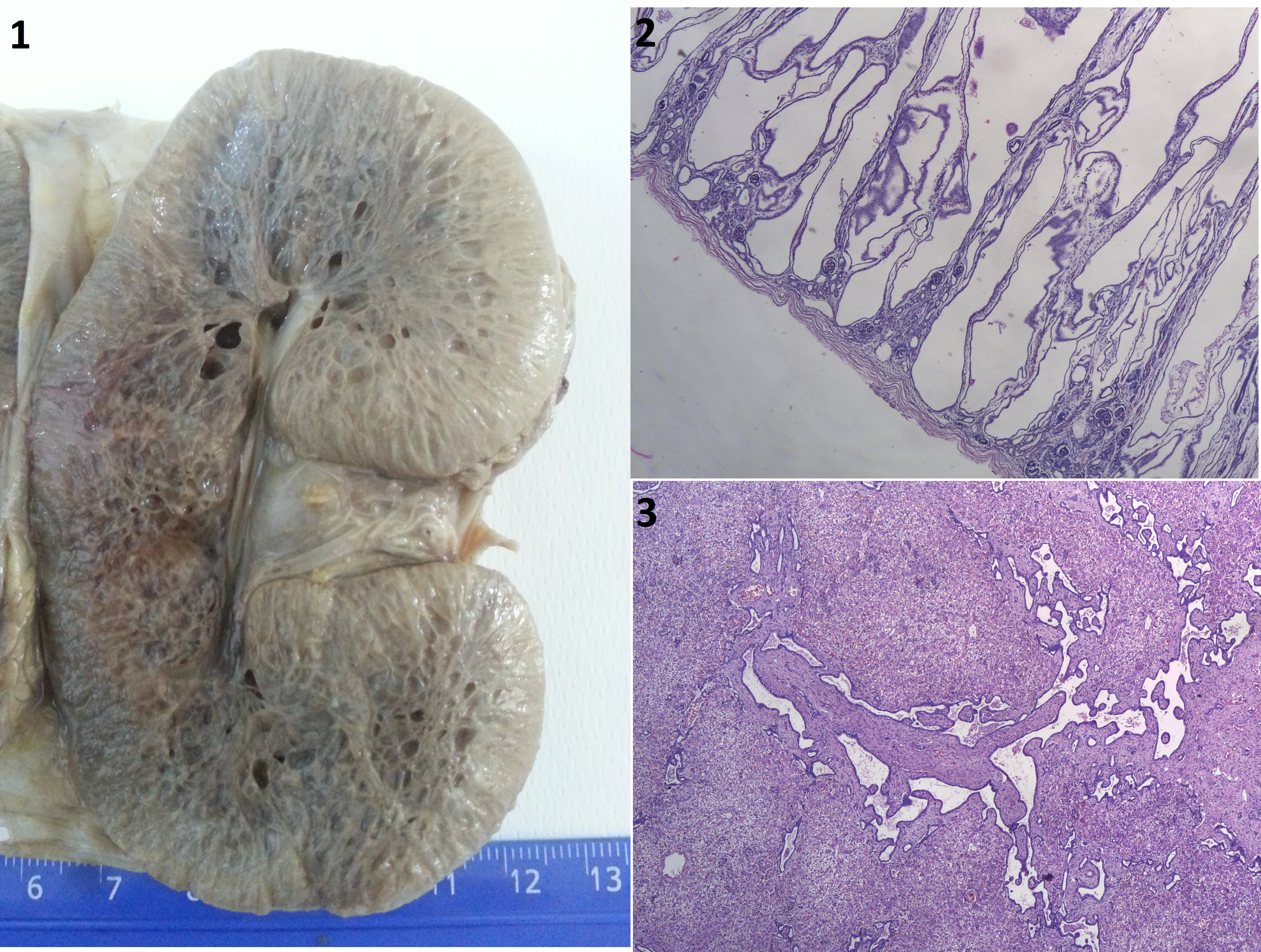
Autosomal recessive polycystic kidney disease (ARPKD) is an important inherited cause of chronic kidney disease with an estimated incidence of 1 in 20,000 live births [1-3]. Mutations of the PKHD1 (polycystic kidney and hepatic disease 1) gene located on chromosome 6p12, are responsible for the entire spectrum of ARPKD [2]. Almost 40% of all affected cases are either stillborn or will die in the neonatal period, due to respiratory insufficiency or kidney failure. In most cases, this disease is accompanied by congenital hepatic fibrocystic lesions (specifical malformation of the liver plate - Meyenburg complex) or by Caroli syndrome [3]. We report a case of a newborn with ARPKD who died within 2 days after birth. An autopsy was performed in the Department of Pathology of the University Emergency Hospital in Bucharest. After a thorough, we established the diagnosis of ARPKD based on specific histological features of this disease, which, as in our case, may establish the final diagnosis without further need for genetic testing. Gross examination revealed hugely enlarged kidneys with cystically dilated collecting ducts that replaced the renal parenchyma almost completely (Figure1). The lungs were mildly hypoplastic. Microscopic examination confirmed the suspicion and showed typical features of ARPKD: numerous radially elongated cysts lined by cuboidal epithelium, decreased renal parenchyma and few remaining rudimentary glomeruli (Figure2). The liver showed dilated portal spaces, with multiple irregularly branching bile ducts and extensive fibrosis consistent with Meyenburg complex (Figure3), classically associated with polycystic liver disease.